There are hits, thank you. (since 1999-06-08)
Calculating formula weight and percent of elements must be the most elemental part in chemical education. However, there are few computer programs, especially running on unix system. Then I have made it.
This program calculate formula weight and percent of each elements for the given chemical formula.
This program was developed on the unix system (FreeBSD 2.2.8R + Motif 2.0), and was confermed to work on IRIX 6.5 system. Later, we changed the development machine to FreeBSD 3.2R + GTK+ 1.2.2. Today, it works well on the FreeBSD 4.2R + open-motif-2.1.30 / gtk-1.2.8 environment ! Though it is a X11 application with GUI [*1], you will be able to use this program through command-line with argument of chemical formula.
[*1] Motif or GTK+ widget sets are required.
Masao KAWAMURA (kawamura@mlb.co.jp) has kindly made an RPM and an SRPM packages for Linux users [*2]. A `spec' file for them was also donated, which is now included in my tarball. Thank you very much, Masao.
[*2] The RPM package given below can be used for the `rpm version 4',
where, for example, MLD5 distribution was assumed.
So, it will not work on the `rpm version 3'-based systems, such as MLD4.
Of course, xmolwt shall be available by compliling from SRPM on the all
other Linux distributions.
URL:http://www.mlb.co.jp/linux/science/
When you type, for Motif version,
% xmolwt
or, for GTK+ version,
% gmolwt
to run the program, calculator's window with GUI is appeared.

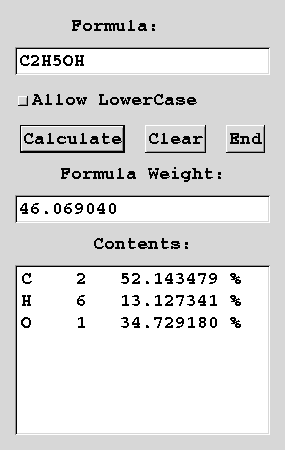
Figure: Molecular weight and elemental analysis calculator.
(Left) Motif version, (right) GTK+ version.
First, input the chemical formula which is constructed by the elemental symbols and numbers at the text field on the top of the window. The atomic groups with nested braket, such as (CH3)2CO, are available as the usual meanings. White spaces are available to separate the elemental symbols.
You can use lower-case in such elemental symbols as h2o, if there are no ambiguity in the given chemical formula. In order to avoid that ambiguity, a white space can be used as a delimiter. (`si' means silicon, while `s i' is sulfur and iodine) In this case, push [Allow LowerCase] button.
After you input the chemical formula, push [Calculate] buttan. Then the formula weight and percent of each elements are printed out.
When you push [Clear] button, the input/output frames will be cleared.
Push [End] button, then the program ends.
X11 has the mechanism about GUI resources, which easily changes the look and feel of the programs.
When you use the Motif version, xmolwt, put an Xmolwt file on your home directry or add it to .Xdefaults/.Xresources.
While you use the GTK+ version, gmolwt, add a dot.gtkrc to .gtkrc on your home directry.
When you use this program at command-line, type as
% molwt <chemical formula>
which is link to GUI version program,
where <chemical formula> is a chemical formula itself,
which contains elemental symbols and numbers of atoms.
For example of ethanol, type
% molwt C2H5OH
Use upper-case and lower-case to write the elemental symbol correctly.
You can use white space to separate the elemental symbols and
can use brakets () to express some functional groups,
where " or ' are required by means of the unix shell argument.
% molwt "Cl C2 H4 OH"
Then, the calculated results of formula weight and percent of each elements
are printed out as follows:
Formula Weight = 80.513500
C 2 29.835245 %
Cl 1 44.033609 %
H 5 6.259447 %
O 1 19.871699 %
And the second example is as follows.
% molwt "(C H3)2 CO"
Formula Weight = 58.079140
C 3 62.039658 %
H 6 10.412757 %
O 1 27.547584 %
Copyright (c), 1998-2001, Ryo MIYAMOTO, All rights reserved.
Re-distribution is permitted, if all source codes and documents are
included in the packed archive.
It is appreciated for me to send any bug-reports, effective patches,
and modification ideas.
Comments for this program are also welcome.
Please take the latest archive (tar + gzip) from HERE.
Ryo MIYAMOTO, Dr.
Molecular Design Engineering,
Department of Materials Science and Technology,
Faculty of Science and Technology,
Hirosaki University
Bunkyo-cho 3, Hirosaki, 036-8561
JAPAN
TEL +81-172-39-3564
FAX +81-172-39-3541
Email rmiya@cc.hirosaki-u.ac.jp