
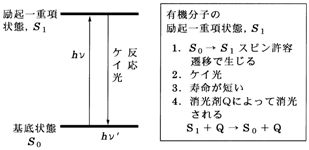
4 �@��N��Ԃ��̌�i�����ߒ��j
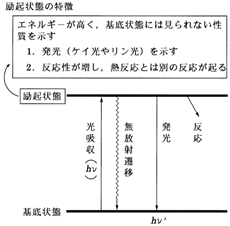
1�D�����ߒ��̎���@���悢��{�_�ł��B���z���Ő�������N��Ԃ͈���ł͂���܂���C�����ꂻ�̃G�l���M�[�������܂��B���̉ߒ��i�����ߒ� deactivation process�j���������I�ߒ� photophysical process�������w�I�ߒ� photochemical process��2�ɑ�ʂ���܂��i�}5�`6�j�B��҂͉��w�ω����ߒ��i�������j���w���C�O�҂͂���ȊO�̉ߒ����w���Ă��܂����C����͂���������ˉߒ� nonradiative process�����ˉߒ� radiative process�ɕ������܂��B�����w�����ߒ�����ɂ��̂悤��3�̉ߒ��i���ˉߒ��C�����ˉߒ��C�����w�I�ߒ��j�������ƍl���Ă悢�ł��傤�B�ȉ������ɂ��ď��ɏq�ׂĂ����܂��B
�@(a) ���ˉߒ��F��N��Ԃ�����Ԃɖ߂�Ƃ��Ɍ�����o����ꍇ�������܂��B��ʂɕ���������o���ꂽ�������� emission�Ƃ����C����ɂ͂��ׂĂ̕������牷�x�ɉ����ĕ��o������M
�}5�@�������I�ߒ��ƌ����w�I�ߒ�
�����i���邢�����x�����j�����~�l�b�Z���X luminescence�i����Ƃ��Ă��j������܂����O�҂͗����C�����ł͌�҂������Ƃ��Ď��グ�邱�Ƃɂ��܂��B���ˉߒ��ɂ����R���o���U�����o�̓��ނ�����i�}1�j�C���̂������R���o�ɂ̓P�C���C������������܂��B������
�@�P�C�� fluorescence�̓X�s�����d�x������2�̏�ԊԂ̕��ˑJ�ځC
�@������ phosphorescence�̓X�s�����d�x���قȂ�2�̏�ԊԂ̕��ˑJ���ƒ�`����Ă��܂��B
�@�L�@���q�œT�^�I�ȃP�C���͗�N��d�����S1 �� �����S0�C�������͍Œ�O�d�����T1 �� �����S0�ւ̕��ˑJ�ڂɊ�Â����̂ł��B
�@�N����(�V)���̂̏ꍇ�C��N�l�d�����4T2g �� �����4A2g���ˑJ�ڂ̓P�C���C�Œ��d�����2Eg �� �����4A2g���ˑJ�ڂ̓������ł��B
�@S1, T1��4T2g, 2Eg�̂悤�ɔ�����������Ԃ�����������邢���������ƌĂ�C������A�����邱�Ƃ͊�{�I�ɏd�v�Ȃ��Ƃ���ɂȂ�܂��B
�@�����ɂ͂��̂ق��ɔ�����ɂ���ē��ɖ��̂��t���ꍇ������C�Ⴆ�G�L�V�}�[�P�C���C�G�L�V�v���b�N�X�P�C���Ȃǂ�����܂��B�G�L�V�}�[�C�G�L�V�v���b�N�X�ɂ��Ă͌�q���܂��B
�}6�@��N��ԂƎ����ߒ�
�@�@�@�@�@�@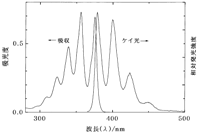
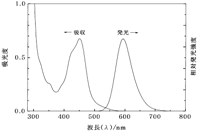
�}7 �A���g���Z���̋z������уP�C���X�y�N�g�� �}8 [Ru(bpy)3]2+�̋z������є����X�y�N�g��
�@�}7�ɗL�@���q�̗�Ƃ��ăA���g���Z���C�}8�ɋ������̗̂�Ƃ��� [Ru(bpy)3]2+�̋z������уP�C���X�y�N�g�������ꂼ�ꎦ���܂����B���̏����̉��ł͋z���Ɣ����X�y�N�g���̌`�������Ԃɒu�������̂悤�Ɍ݂��ɑΏ̂ɂȂ邱�Ƃ��m���Ă��܂��B����������W�Ƃ����C�A���g���Z���̏ꍇ�ɗǂ����藧���Ă��邱�Ƃ�������܂��B
�@�x���P�C��delayed fluorescence�́C��N��d�����S1 �� �Œ�O�d�����T1�ɖ����ˑJ�ڂ�����Ă�S1�ɖ߂�C��������P�C�����o���ꍇ�ł��i��q�}10�j�B���̖��͈�U�O�d�����o�R���邽�ߎ��ԓI�ɒx��邱�ƂɗR�����Ă��܂��BS1���猩����P�C���ł�����C�X�y�N�g���̌`�͒ʏ�̃P�C���ƑS�������ł��B
�@����U�����o�͗�N��Ԃ����̎h�����Č�����o����ꍇ�ŁC���[�U�[�͂���Ɋ�Â��Ă��܂����C���̂��߂ɂ͈��̏������K�v�ł���ʏ�̏������ł͌����܂���B�����Əڂ�����4-4-e���Q�Ƃ��ĉ������B
�@(b) �����ˉߒ��F��N��Ԃ�����Ԃɖ߂�Ƃ��Ɍ�����o���Ȃ��ꍇ�������C�G�l���M�[�͔M�I�Ɏ��̗͂n�}�ɗ^�����܂��B���̂����X�s�����d�x�������d�q��N��ԊԂ̖����ˑJ�ڂ������]�� internal conversion�Ƃ����܂��B�Ȃ������ˉߒ��͗��_�I�Ȏ�舵�����Ȃ���Ă�������ł��������ł͏q�ׂ܂���B
�@���Ԍ������邢�͌n�Ԍ��� intersystem crossing�F�����ˉߒ��̒��œ��M���ׂ����̉ߒ��́C�X�s�����d�x���قȂ�2�̏�ԊԂ������ˑJ���̂��Ƃ������܂��B��\�I�ȗ�ɗ�N��d��S1 �� �Œ�O�d��T1�ւ̃X�s�����J�ڂ�����܂��B�Œ�O�d����Ԃ͌����w�ő傫�ȈӋ`�������C������3-4-c�ŏq�ׂ��悤�ɍŒ�O�d����Ԃ͒��ڋz���ł͐����Ȃ��̂�S1���疳���ˑJ�ڂŐ�����ȊO�ɂ���܂���B�]���č��Ԍ����͋ɂ߂ďd�v�ȉߒ��ł�����CT1���ǂ̈ʂ̊����Ő����邩��S-T�J�ڊm���ƌĂ�C���̒l�����߂邱�Ƃ��傫�ȊS���ƂȂ��Ă��܂��B
�@S1 �� T1�X�s�����J�ڂ́C�d���q�����݂���Ƃ��̋������������邱�Ƃ����_�I�ɂ������I�ɂ��m������Ă��܂��B������d���q�����Ƃ����C�d���q�����q�����ɂ���ꍇ�i�����d���q�����j�Əd���q����O�����Ƃ��ēY�������ꍇ�i�O���d���q�����j�̓��ނ�����܂��B
�@(c) �����w�I�ߒ��F��N��Ԃ����w�������s���ߒ��ŁC���̔����͔M�I�ɋN���锽���Ƌ�ʂ��Ĉ�ʂ������w�����ƌĂ�Ă��܂��B�G�l���M�[�I�ɍ�����Ԃ���̔����ł�����C����Ԃ�蔽�����������C�܂�����ԂƂ͈قȂ锽�����N����̂�����������܂���B�L�@�����w���i�W���Ă����ő�̗��R�������ɂ���ł��傤�B�����̏ꍇ�C�������ԑ̂Ƃ��ă��W�J����C���̂ł͌ܔz�ʒ��ԑ́C���z�ʒ��ԑ̓����܂��B
2�D��{�p���@�����w�ł̏�p�������m���Ă��������܂��傤�B
(a) �ʎq���ʂ��邢�͗ʎq���� quantum yield�F��N��Ԃ���ɏq�ׂ��悤�ȃG�l���M�[�������e�ߒ����C1�̌��ʎq������ǂ̈ʂ̊����ōs�������������l�ł��B�e�ߒ��ɑ��āC�m���ܖ��ɂ��Ă���ߒ��ɐi�ޕ��q�̐��n�^�m�����ɋz�����ꂽ���ʎq�����Ȃ킿���z����Iabs�n�Œ�`����܂��B�P�C���̗ʎq���ʁC�����̗ʎq���ʂȂǂƓ���̉ߒ�������K�v������܂��B�Ⴆ�P�C���̗ʎq���ʃ�F��0.5�Ƃ͗�N��Ԃ�50�����P�C�����o�����Ƃ��Ӗ����܂��B
�@(b) ���� lifetime�F��N��Ԃ���̕��˂���і����ˑ��x�萔�����ꂼ��ke, knr�Ƃ��C�����̑��x�萔��kr�Ƃ���Ƃ��C
��m�i����������邢�����ώ����j��1�^(ke + knr + kr)�@�@(10)
��0�i���R�������邢�����ˎ����j��1�^ke�@�@(11)
�Ƃ����܂��B
�@�L�@���q�̗�N��d��S1�̂悤�Ɋ���Ԃւ̑J�ڂ��X�s�����e�̂Ƃ��́C����Ԃɑ����߂邽�߂�S1�̎����͒Z���C����L�@���q�̍Œ�O�d��T1�̓X�s�����J�ڂł��邽�߂ɂ��̎����͒����Ȃ�܂��B��ʂɗL�@���q�ŃP�C���������Z���C�����������������̂͂��̂��߂ł��B
�@(c) �ʎq���ʂƎ����F�����̉ߒ��ɑ���ʎq���ʃ��Ƒ��x�萔k�̊Ԃɂ͎��̊W������܂��B
����k��m�@�@(12)
�Ⴆ�P�C���̗ʎq���ʂ���ё��x�萔�����ꂼ�ꃳF, kf, �������̗ʎq���ʂ���ё��x�萔�����ꂼ�ꃳR, kr�Ƃ����
��F��kf��m�@�@(13)
��R��kr��m�@�@(14)
�@(d) ���w���ʌv chemical actinometer�F�ʎq���ʂ����߂�̂ɕK�v�Ȍ��z���ʂ��C�����w�����𗘗p���đ��肷����@�ŁC���ۂɂ͎g�p��������̋����𑪒肵�܂��B���Ȃ킿�����w�����̗ʎq���ʂ����m�̕W�����������ۂɌ��Ǝ˂��Ĕ��������C���̔����ʂ�������̋���������ɂ͌��z���ʂ��t�ɋ��߂܂��B���ʌv�Ƃ��邩��Ƃ����ĕ����I�ȃ��[�^�[��A�z���Ă͂����܂���B�ǂ��g������ʌv�Ƀg���I�L�T���g�S(�V)�_�J���E���C���C�l�b�P���Ȃǂ�����܂��B
3�D�d�v�Ȍ������I�ߒ��@�������I�ߒ���ʂ��Ċ�{�ƂȂ�_���q�ׂ܂��B�@
(a) �t�����N�E�R���h���̌����F���̌����́C�u�d�q�J��(10-15 s)�͊j�̉^��(10-12 s)�ɔ�ׂ�Ɣ��ɑ����̂ŁC�d�q�J�ڂɍۂ��Ċj�݂͌��̈ʒu���邢�͉^���G�l���M�[������Ȃɕς��Ȃ��v�Ƃ������̂ł��B�j�͓d�q������1840�{�d���ł����瓮�����݂��C�d�q���J�ڂ���Z�����Ԃ̊Ԃ͓����Ȃ��Ƃ݂Ȃ��Ă��悢���낤�C�Ƃ������Ƃł��B

�}9�@�t�����N�E�R���h���̌����@
�@���̌����͓d�q�J�ڂ𗝉�����̂ɏd�v�ł��邱�Ƃ�}9�ōl���Ă݂܂��傤�B����Ԃ̐U������v��i �����N��Ԃ̐U������v��j �ւ̑J�ڂ���ʂ�i �� j�J���Ƃ����܂��B�}��(1)�͊���ԂƗ�N��ԂƂň���Ȋj�z�u�����܂�ς��Ȃ��ꍇ�ł��B����Ԃ�v��0����z�����N����Ƃ��C�J��b(0 �� 3�J��)�͊���Ԃɔ�ׂăG�l���M�[���������邽�߂ɁC�܂��J��c(0 �� 0�J��)�͈ʒu���������邽�߂ɁC���ꂼ��J�ڂ���m���i�J�ڊm���j���Ⴍ���x���ア���ƂɂȂ�܂��B�]���Đ}�ł͑J��a(0 �� 0�J��)���ł��J�ڂ��₷���Ɨ\�z�ł��܂��B���̂��Ƃ͂��̐}�ł͕���ɂ����̂ł����C�|�e���V�����G�l���M�[�Ȑ��ƌĂ��Ȑ��ōl����Ɩ��炩�ƂȂ�܂��B���(2)�ł́C��N��Ԃ̈���Ȋj�z�u������Ԃ����������Ă��邽�߂ɍł��J�ڂ��₷���̂�0 �� 3�J�ڂł��邱�Ƃ������Ă��܂��B���̂悤�ɁC�d�q�J�ڂ͊j�̈ʒu��G�l���M�[���傫���ς��Ȃ����̂��������̂ŁC�ł��m���̍�������Ԃ���̑J�ڂ͐����̖��N��Ԃ̃|�e���V�����G�l���M�[�Ȑ��ƌ���鏊�ł���ƌ��_����܂��i�����J���ƕ\������܂��j�B�����łȂ��J�ڂ̊m���͒Ⴂ�̂ł��B
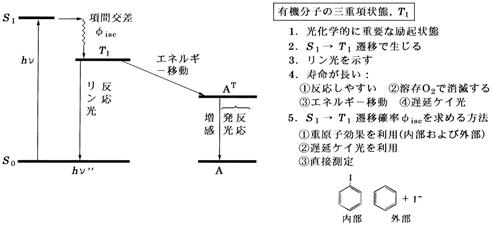
(b) Jablonski �_�C�A�O�����F�t�����N�E�R���h���̌����Ɋ�Â��ė�N��ԊԂ��邢�͗�N���-����ԊԂ̓d�q�J�ڂ��U�����ʂ��܂߂ĕ\�킷�_�C�A�O�����̂��Ƃ������܂��B���}10�Ɏ����܂����B
�@����Ԃ��琂���J�ڂŗ�N��ԂɒB��������̐U�����ʁi�t�����N�E�R���h������j��v��0�ł͂Ȃ���Ԃ́C�G�l���M�[�I�ɍ����U����Ԃł����ė�N��ԂɂƂ��Ĉ���ł͂���܂���B�}�Ńt�����N�E�R���h����Ԃ�S1(v��3)�ł���C�]���Ĉ���ł͂Ȃ��̂ł₪�ĕ��q�͐U�������Ȃ���G�l���M�[�������C�ŏI�I�ɐU������v��0�̏��ɗ��������܂��B���ꂪ�U���ɘa vibrational relaxation�ƌĂ��ߒ��ŁCS2 �� S1, T2 �� T1�Ȃǂ̓����]���ɔ�ׂĔ��ɑ����̂ł��̉ߒ��͗D��I�ɋN���邱�Ƃ��m���Ă��܂��B
�@�}10�Řb���܂Ƃ߂Ă݂܂��傤�B�t�����N�E�R���h���̌����ɂ���ė�N��d�����S1(v��3)�̃t�����N�E�R���h����Ԃɗ�N���ꂽ���q�́C�U���ɘa�ɂ���đ��₩�Ɉ����S1(v��0)�̏��ʂɒB���C��������P�C�����o������C�������s������C�����˓I�Ɋ���Ԃɗ�������C

�}10�@Jablonski�_�C�A�O�����@
���邢�͍��Ԍ����ɂ���čŒ�O�d���ɍs���Ȃǂ̏����ߒ����s���̂ł��B������S1 �� T1���Ԍ����Œ��ӂ��ׂ��́CT1�ɑJ�ڒ���̓t�����N�E�R���h���̌����ɂ����T1�̒��ŐU���I�ɍ������ʁi�}�ł�v��3�j�ɂ���C�Œ�O�d���ɂƂ��Ă͂�͂����ł͂Ȃ����Ƃł��B�]����S1�̂Ƃ��Ɠ��l�ɐU���ɘa�ɂ���đ��₩�Ɉ����T1(v��0)���ʂɒB������C���������o������C�����ˑJ�ڂ��s���킯�ł��B���ɂ͉��炩�̕��@�ŃG�l���M�[���l�����CT1(v��3)�ɒB������ɋt���Ԍ����ɂ���čĂ�S1�ɖ߂�ꍇ������܂��B���ꂪ�O�q���x���P�C���ł��B���̗Ⴉ�番��悤�ɁC2�̗�N���S1-T1�Ԃ̍��Ԍ����̓_�C�A�O������Ő������ŕ\�킳��܂��B
�@�P�C���͐U���ɘa�̌��ʁCS1�̒��ŃG�l���M�[�I�ɍł��Ⴂ v��0����N����܂��B���̂��߃P�C���X�y�N�g���̋ɑ�g���̓t�����N�E�R���h�����S1(v��3)�Ƃ̃G�l���M�[���̕������z���X�y�N�g���̋ɑ�g���������g�����Ɍ�����̂����ʂł��B���̂悤�ȋz���ƃP�C���Ƃ̃G�l���M�[�����X�g�[�N�X�V�t�g Stokes shift�Ƃ����C��N��Ԃ̊j�z�u������Ԃ����������Ă���قǃV�t�g�͑傫���Ȃ�܂��B
�@�U�����ʂ��l�����ꍇ�C�z���ł�i �� j �J�ڂ��C�܂������ł͍Œ�̐U�����ʂ����0 �� i �J�ڂɊ�Â��X�y�N�g�����d�q�J�ڂɔ����Č����܂��B������U���X�y�N�g���Ƃ����C���ꂪ���ꂽ�X�y�N�g���̌`�E�l�q���U���\���Ƃ����܂��B�}7�Ɏ������A���g���Z���̋z���C�P�C���X�y�N�g���ɂ͂��ꂪ�m�R�M����ɖ��Ăɂ݂��邱�Ƃ�������ł��傤�B
4�D�q�ߒ��@����܂ł͗�N���q���P�Ƃɍs���ߒ����q�ׂ܂����B��������N���q�͑��̕��q���邢�͌��q�C�C�I���Ƃ̊Ԃɋ�������q�ߒ����s���܂��B�@
�@(a) �����ƃG�l���M�[�ړ��F�����N��d��S1������(15)�`(18)�܂ł̎����ߒ����s���Ƃ��܂��B���̂悤�Ȉ�A�̉ߒ����X�L�[�� scheme�Ƃ����܂��B
�@�X�L�[��(15)�`(17)�ɑ��ė^�������̂͊��ɏq�ׂ����̂ňꕪ�q�ߒ��ł����C�ߒ�(18)��S1���O���̕��q�i��O�����jQ�ƏՓ˂��Ċ����S0�ɖ߂�q�ߒ��i�����j�ł��B����������Ƃ����CQ�������� quencher�Ƃ����܂��BS1��Q�ɂ���ď������ꂽ���ƂɂȂ�܂��B
S0 �� S1 ���z��(h��)�@�@(15)
S1 �� S0 + h��' kf �P�C��(�� > ��')�@�@(16)
S1 �� S0 knr �����ˑJ�ځi�����]���j�@�@(17)
S1 + Q �� S0 + Q kq ���� quenching�@�@(18)
�@(18)�ł͏����̌���Q�ɕω��͂���܂��C(19)�̂悤��Q����N��ԂɂȂ�ꍇ������C���̂悤�ȉߒ����G�l���M�[�ړ� energy transfer���邢������ sensitization�Ƃ����܂��B�G�l���M�[�ړ��̖��O�́C�G�l���M�[�������Ă�����������Ă��Ȃ����փG�l���M�[��^����C�܂�G�l���M�[���ړ����邱�ƂɗR�����Ă��܂��B�O�҂��G�l���M�[���^���ƌĂ�CS1��T1�Ȃǂ�����ɑ������܂��B�����҂̓G�l���M�[�����炤���Ȃ̂��G�l���M�[��e���ƌĂ�܂��B�����Œ��ӂ��Ȃ���Ȃ�Ȃ����Ƃ́C
�@�u�G�l���M�[�ړ��́C�G�l���M�[���ʂ�����������Ⴂ���ւƋN�����v�킯�ł�����C
�@�u�G�l���M�[���^�̂̃G�l���M�[���ʂ��G�l���M�[��e�̂̃G�l���M�[���ʂ��������v
�Ƃ����������K�v�Ȃ��Ƃł��B
�@�����̏ꍇ�C�uS1��Q�������v�Ƃ��C�uQ��S1�ɂ���đ������ꂽ�v�Ƃ������Ȍ����������܂��B�G�l���M�[�ړ��Ő�������N���Q*�́C���ڗ�N�Ő������Ƃ��Ɠ����悤�ɔ�������������(20)�C���������肵�܂�(21)�B�O�҂����������C��҂����������Ƃ��ꂼ��Ăт܂��BS1����N��d���̂Ƃ��C����Q*�͗�N��d���ł���̂������P�C���������܂��B
S1 + Q �� S0 + Q* �������邢���G�l���M�[�ړ��@�@(19)
Q* �� Q0 + h��'' �������� sensitized emission(�� > ��'')�@�@(20)
Q* �� P(������) �������� sensitized reaction�@�@(21)
�@��N��d���i���x��1X����т���Ƃ͈قȂ�1Y�Ƃ���j�������U�镑���̒��ŋ���������̂́C1X�Ɗ����X0���q����ė�N��Ԃ̓�ʑ̂��`�����邱�ƂŁC�ł�����ʑ̂��G�L�V�}�[(excimer��excited dimer�̈Ӗ�)�Ƃ����܂�(22)�B���l�ɓ�ʑ̌`�����݂��ɈقȂ镪�q��1X��Y0����ł����Ƃ��͂��̓�ʑ̂��G�L�V�v���b�N�X(exciplex��excited complex�̈Ӗ�)�Ƃ����܂��B���̍ő�̓����͗��҂Ƃ���N��Ԃł̂ݑ��݂������ƂŁC����Ԃɖ߂�ƕ������Ă��܂��܂��B
1X + X0 ��1X2* �G�L�V�}�[�i��N��ʑ́j�@�@(22)
1X + Y0 �� 1(XY)* �G�L�V�v���b�N�X�@�@(23)
�@�Œ�O�d���͎����������̂œ��l�ȉߒ�����N��d�������N����₷���Ȃ�܂��B�Œ�O�d������̃G�l���M�[�ړ��͂������ʂɌ�����ߒ��ł����C���ɗL�@���q�̏ꍇ�C�Œ�O�d���͗n�t���ɑ��݂����n���_�fO2�ɂ���Đv���ɏ�������C�n���_�f����d���_�f singlet oxygen(1O2*)�ɂȂ邱�Ƃ��m���Ă��܂�(24)�B���ꂪ�O�d���Ɋւ���������s�����߂ɗn���_�f����������i�r�C���邢���E�C�j���s��Ȃ���Ȃ�Ȃ����R�ƂȂ��Ă��܂��B
T1 + O2 �� S0 + 1O2* �����@�@(24)
�@�ߒ�(26)��T-T�������܂ވ�A�̉ߒ�(25)�`(27)�ɂ���āCQ�i���̏ꍇ����e�� acceptor�ƌĂ��������ʁj�̗�N��d��1Q1���猩����P�C���������x���P�C���Ƃ����܂��B
T1 + Q0 �� S0 + 3Q1 �G�l���M�ړ��[�@�@(25)
3Q1 + 3Q1 �� Q0 + 1Q1 T-T���� T-T annihilation�@�@(26)
1Q1 �� Q0 + h��' �����x���P�C���@�@(27)
�@(b) Stern-Volmer�v���b�g�F�O�q�̃X�L�[��(15)�`(18)�ɑ���������Ԗ@photostationary state method��K�p����ƁC�����������܂��B�A�����x�萔�̓X�L�[���Ɏ��������̂Ƃ��܂��B
��F0�^��F ��IF0�^IF
��1 + kq��0[Q]�@�@(28)
��1 + KSV [Q]�@�@(29)
�����Ń�F, IF�͂��ꂼ��S1����̃P�C���̗ʎq���ʂ���уP�C�����x�ł���C��0��1�^(kf + knr)��S1�̎�����\�킵�܂��B���̂Ƃ���t���̃[����[Q]�i������Q�̔Z�x�j���[���̂Ƃ���\�킵�Ă��܂��B�����KSV�i��kq��0�j�ŕ\�킳���ʂ�Stern-Volmer�萔�i�V���e����-�t�H���}�[�萔�j�Ƃ����āC���̒l���傫���قǏ����������悭�N���邱�Ƃ������܂��B

�}11�@Stern-Volmer �v���b�g
�@��(28)���邢��(29)���番��悤�ɁC�c���Ƀ�F0�^��F���邢��IF0�^IF�����C������[Q]������ăv���b�g�����1��ؕЂƂ��C�X��KSV�̒����ɂȂ�܂��i�}11�j�B�����Stern-Volmer�v���b�g�Ƃ����Ĕ����┽���ɑ��Ĉ�ʂɐ��藧�d�v�ȊW�ł��B
�@(c) ��N��Ԃ̗D�ʐ��F��N��Ԃ͑傫�ȃG�l���M�[��L���Ă��܂�����C����Ԃɔ�ׂėD��Ă���_����������C���̌����ȗ�͔����Ɍ����܂��B���Ȃ킿�M�����ł͈�̔�����
�@�@�@�@�@�@�@�@
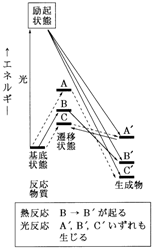
�@�@�}12�@��N��Ԃ���̔������L�������邱�Ƃ������}
���ĉ���ނ��̔����o�H������܂����C���鉷�x�Ŏ��ۂɋN����̂͂��̂����ł��������G�l���M�[�̏����Ȍo�H�����ł��B�����Ƒ傫�Ȋ������G�l���M�[�̔������N�����ɂ͉��x���グ�Ȃ���Ȃ炸�C���ɂ͔M�������N�����Ă��܂������Ȃ����Ƃ��\�����蓾�܂��B�}12�Ɏ�������ł͎O�ʂ�̌o�H�̂����C�o�HA��C' �͊������G�l���M�[���傫���������x�����߂ɁCC��A' �͐�����������łȂ��Ƃ����M�͊w�I�ȗ��R�̂��߂ɁC���ɔ����͋N���ɂ����Ȃ��Ă��܂��B���ۂɋN����̂�B��B' ���������ł��B�������������ł͗�N��Ԃ��\���ȃG�l���M�[�������Ă��邽�߂ɁC�G�l���M�[�s�����痈����͉�������Ă��܂��܂��B���̌��ʁC A' , B' , C' ������̐���������������̂ł��B�����Ƃ�A' �͒����ɕ������Ă��܂��ł��傤���B
�@������D��Ă���_�́C�M�����ł͑傫�ȃG�l���M�[��^����㏞�Ƃ��Ĕ����n�̉��x�������Ȃ�̂ɑ��C���Ǝ˂ł͔����n�̉��x���オ�邱�Ƃ͂Ȃ��Ƃ������Ƃł��B�A�����ۂɂ͌��������v��_�������Ƃ��C�����v���M�����邽�߂ɔ����n�̉��x���オ�邱�Ƃ͂���܂��B�����K���ȕ��@�ł��̔M���������Ƃ��ł���Δ����n�̉��x�͑������Ȃ��͂��ł��B
�}13�@�L�@���q�̗�N��d����Ԃ̂܂Ƃ�
�@�@�@�@�@�@�@�@�@�@�@�@�@
�@�@�@�@�@�@�@�@ �}14�@�L�@���q�̗�N�O�d����Ԃ̂܂Ƃ�
�@(d) ��N��d���C�Œ�O�d����Ԃ̓����F�L�@���q�̗�N��d������эŒ�O�d���͌����w�ɂƂ��Ċ�{�ɂȂ�d�v�ȗ�N��Ԃł��B���܂ŏq�ׂ����Ƃ𒆐S�ɐ������Đ}13�C�}14�Ɏ����Ă����܂�����C�ł��邾���Q�Ƃ��ĉ������B�Ȃ��Ȍ����������Ă����̐}�ŐU�����ʂ͏Ȃ��Ă���܂��B
�@(e) ���[�U�[�F����ɂ��Ă�1-1-f�C4-1-b�Ŋ��ɊȒP�ɐG��܂������C������x�̒m����~�������C�����ł�����x�U��Ԃ��Ă݂����Ǝv���܂��B
�@���[�U�[��light amplification by stimulated emission of radiation�i�U�����o�ɂ��������j�̓������������laser�̈Ӗ��ł��B�}1�Ɏ������悤�ɁC���q�������z�����Đ���������N��Ԃ͎��R���o�ɂ���Ĕ����i���̏ꍇ�P�C���Ƃ��Ă����܂��j���o���̂����ʂł��B�������A�C���V���^�C���͂��̗�N��Ԃ����̏�ɂ���Ƃ��C���̌��̎h�����ē��ˌ��Ɠ����ʑ��C�U������������1�̌����C�P�C���Ƃ͕ʂɁC���o����ƍl�����̂ł��B���ꂪ���ɂ���ėU������ďo�������C�܂�U�����i�����[�U�[�j�ł����B
�@�ʏ�̏������ł��{���c�}�����z�ɂ���Ċ���Ԃɂ��镪�q�������|�I�ɑ������߂ɁC��N��Ԃ���o��͕̂��ʂ̃P�C�������ł��B�U�����o�����Ȃ킿���[�U�[���������邽�߂ɂ͎���3�̏��������������K�v������܂��B�@���[�U�[������o�������[�U�[�}�������鎖�C�A����������������C�B���]���z���������Ă��鎖�ł��B���̂����A���s�����߂Ɏg����̂����[�U�[���U��i�}15�j�ł��B2�̔��ˋ��˖ʂ����������悤�ɕ��ׁC����̋��͌�
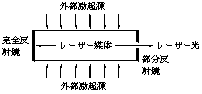
�}15�@���U�� �}16�@���[�U�[�̔��U
�����S�ɁC��������͕����I�ɔ��˂���悤�ɂ��Ă����܂��B�O����N��������Ǝ˂���ƁC���[�U�[�}������o�����R���o���i�P�C���j�͗����̋��̊Ԃ��������܂��B���̊Ԃɕʂ̕��q�ɓ��˂��邲�Ƃ�1�̗U�����������o���C�v2�̌��ƂȂ�̂Ō��̐��͎���ɑ������ƂɂȂ�܂��B�ŏ��͗U�����̌����͑����Ă��炸�C���ɕ��s�łȂ����͌n�O�ɏo�Ă����܂����C���ԂƋ��Ɍ��������������ƂȂ��Ă����܂��B�����Ă�����x�܂Ō������߂�ꂽ�Ƃ������̋����烌�[�U�[���Ƃ��ĕ��o�����d�g�݂ɂȂ��Ă���̂ł��B
���]���z�͗�N��Ԃɂ��镪�q��������Ԃ����������邱�Ƃł����C���ꂪ�ǂ̂悤�Ɏ�������Ă���̂����������̂��}17�ł��B�����悭�������邽�߂ɓs���̂悢���[�U�[�}����I��ł���CE1, E2, E3��3�̏��ʂ���Ȃ�̂�3���ʃ��[�U�[�ƌĂ�Ă��܂��B
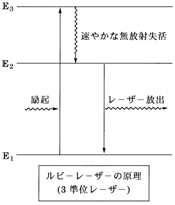
�}17�@3���ʃ��[�U�[�̌���
���܌���N�ɂ���Ċ����E1����E3�ɗ�N���܂��ƁC������E3��E2��E1�Ə��ɑ��₩�ɋN���邱�ƂɂȂ�C���̂܂܂ł͔��]���z�͎������܂���B����������E3��E2�����ˑJ�ڂ����₩�ɋN��CE2��E1�����ˑJ�ڂ��x����CE2�̕��q��N2��E1�̕��q��N1��葽���iN2 ��N1�j�Ȃ���E2��E1�̊Ԃɔ��]���z���������CE2����̃��[�U�[���o���N���邱�ƂɂȂ�܂��B3�̏��ʊԂ̖����ˑJ�ڂ��������x�����̓��[�U�[�}���̐����Ɉˑ����܂�����C���炩���߂����̏�����Ă���}����I�ԕK�v������܂��B
�@�ŏ��Ɏ��������U�����o���̓}�C�N���g�ł����ă��[�U�[�ƌĂ�C�����Ɣg���̒Z�����̈�̌��C���[�U�[�ł͂���܂���ł����B���̃��[�U�[�͌ő̃��r�[����̂��̂ł������C���̔}���ƂȂ����̂̓��r�[�ɔ��ʊ܂܂��Cr3+�ł����B�����E1��4A2g, E2��2Eg, E3��4T2g�ɑ�������3���ʃ��[�U�[�ł��B2Eg �� 4A2g�i����ԁj���X�s�����J�ڂŒx���̂ŁC2Eg�̕��q���������Ȃ邱�Ƃɂ����2Eg�̕��q�� �� 4A2g�̕��q���ƂȂ�C���҂̊Ԃɔ��]���z����������2Eg����̃��[�U�[���o���N����̂ł��B
�@�����ă��[�U�[�}����I�ԍۂɕK�v�������̏���^�����̂������w�ł������킯�ŁC���ꂪ�\�ł������̂������̊�b�I�ȃf�[�^�̒~�ς����������������Ƌ������Ă����܂��傤�B